CHM621
Chemical Binding
Procedure
to run gaussian 94 in the CC
Server name : e10k-1
IP address : 172.31.1.91
To run gaussian 94 you need to set up the environment in your login, that will enable you to access the software. The set up parameters of your login are usually given in the a file named .profile. One needs to edit the file and make the changes needed to run the software. Follow the instructions below to do so.
telnet e10k-1
login: (type your id)
passwd: (your passwd )
$ vi .profile
Inside the .profile add the following lines at the end of the file. Go to the
end of the file and type 'o'. This will make the cursor to the next line where
you should type the following lines.
export
GAUSS_EXEDIR=/extra/local/sm/gaussian/g94
export
BASIS=/extra/local/sm/gaussian/g94/basis
export GAUSS_SCRDIR=/tmp
alias
g94='/extra/local/sm/gaussian/g94/g94'
After typing the above, press 'ESC' key to come out of the insert mode and then
save and quit the file by :wq
Now execute the .profile to activate the new settings by:
$ . .profile
Check if gaussian 94 is working by typing
$ g94
You will get :
Entering Gaussian System, Link 0=/extra/local/sm/gaussian/g94/g94
Kill the process by pressing Control-C.
Now you have set up your login to run gaussian in e10k-1.
----------------------------
|
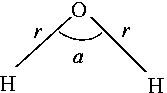
Sample input file for
water |
|
$ vi h2o.com
This will open a file called h2o.com. To type inside the file, you have to go
to the insert mode by typing 'i'.
Now type the following:
|
|
General format of input file:
|
After typing the input file, press 'ESC' key to come out of the insert mode and
then save and quit the file by :wq
Now h2o.com is the input file to run gaussian 94.
---------------
Running gaussian
To run gaussian g94 type the following:
$ g94 h2o.com &
This will take some time. After the execution, the program will generate an
output file called h2o.log. Go through the h2o.log file. It contains all
the information the program has calculated and the details of the execution.
The summary of the results are at the end of the file.
Summary:
Final structure in terms of initial Z-matrix:
O
H,1,r1
H,1,r1,2,a1
Variables:
r1=0.98931002
a1=100.03447411
Test job not archived.
1\1\GINC-E10K-1\FOpt\RHF\STO-3G\H2O1\LOURDU\09-Oct-1903\0\\#HF/STO-3G
TEST POP=REG FOPT\\test h2o\\0,1\O,-0.1008650707,0.,-0.0773964909\H,-0
.0580093199,0.,0.9109848586\H,0.8649298855,0.,-0.2918129315\\Version=S
un-SVR4-Unix-G94RevC.2\State=1-A1\HF=-74.9659012\RMSD=3.919e-09\RMSF=7
.648e-05\Dipole=0.533524,0.,0.4093874\PG=C02V [C2(O1),SGV(H2)]\\@
....
....
...
Job cpu time: 0 days 0 hours 0 minutes 20.2 seconds.
File lengths (MBytes): RWF= 5
Int= 0 D2E= 0 Chk= 1
Scr= 1
Normal termination of Gaussian 94
HF=-74.9659012 is the Hartree Fock optimized energy for water.
The last line should be "Normal termination of Gaussian 94" for an
error free execution.
-----------
Important note :
During the execution of the gaussian program, a lot of scratch (temperory)
files are generated. These files are set up in such a way that they are written
in the /tmp directory of the server (recollect export GAUSS_SCRDIR=/tmp
in the .profile). When the program terminates without errors, these
scratch files are automatically removed. But when the program terminates with
some error, these files are not removed. Hence it
is your responsibility to remove these scratch files, as these are
very large files usually ranging from 500 MB to 2 GB depending on the method of
calculation and basis set. Please remove these files to let others use
the /tmp directory efficiently. To remove these files type :
$ rm /tmp/g94*
Give 'yes' for all the files to remove them. Only those files that belong
to you will be removed.
A file 'core' will also be generated in the working directory, if the program
terminates with errors. Do remove it, as it is a large unwanted(!!) file.
-----------
Queries : ramcn@iitk.ac.in