Overview
Research in Chatterjee group focuses on the development of accurate multiscale models, such as continuum mesoscopic equations, accelerated kinetic Monte Carlo, accelerated molecular dynamics methods and DFT. We are employing these models to investigate materials for batteries, fuel cells, opto-electronic devices and heterogeneous catalysis.
[+] Click topic to expand
Continuum mesoscopic equations
Phenomenological equations such as Fick's law are sometimes inaccurate when atoms interact with each other. In such cases, it is important to correctly build the atomic interactions into the continuum model. Continuum mesoscopic equations are obtained from molecular scale descriptions via spatial and temporal coarse-graining. Examples of nanostructures growth studied using mesoscopic equations are shown below.

Accelerated superbasin KMC
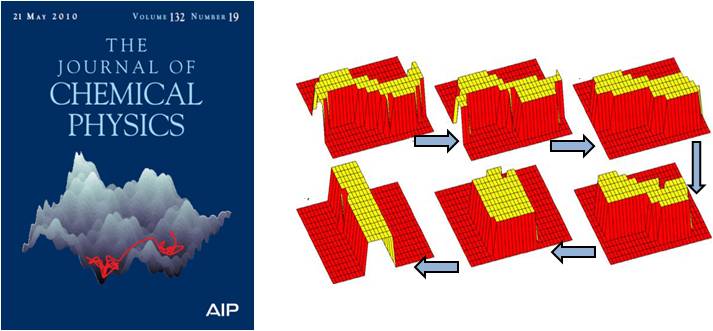
The time-step in kinetic Monte Carlo (KMC) method goes inversely as the sum of the process rate constants for the current state of the system. Whenever the system is trapped in a collection of states connected via fast atomic processes, KMC will keep sampling these processes and the system remains trapped in the superbasin for a large number of KMC iterations. The accelerated superbasin KMC method modifies the underlying potential energy surface allowing the system to escape superbasin more quickly thus accelerating the KMC dynamics. Figure below is from the cover of Journal of Chemical Physics highlighting our work. An example of a shape transformation of a nanowire is as shown below.
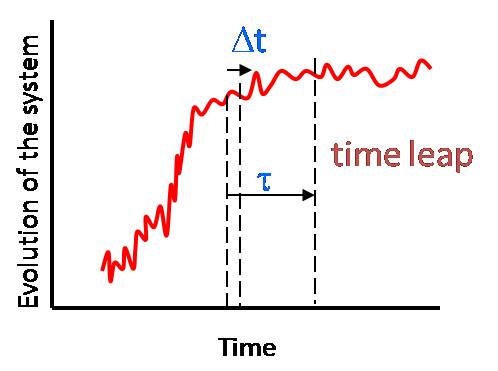
Tau-leap KMC methods
In standard kinetic Monte Carlo (KMC) method, one atomic process is randomly selected in each iteration. The tau-leap KMC method allows one to randomly select several KMC processes in a single time-step that is much larger than standard KMC thus allowing KMC to reach longer times in fewer number of iterations.
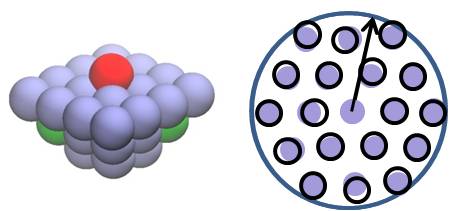
Local environment KMC
Traditionally KMC methods have employed the lattice approximation, where atoms are assumed to reside on lattice sites. However, this approximation cannot be employed in situations where atoms can reside in off-lattice positions, e.g., with multicomponent materials or when elastic strain is present. In such cases, the local environment KMC (LE-KMC) method can be used. LE-KMC is a self learning, off-lattice KMC method, i.e., it learns about the types of processes that can occur in a material system in terms of the local atomic environment as the dynamics of the system is evolved (see below).
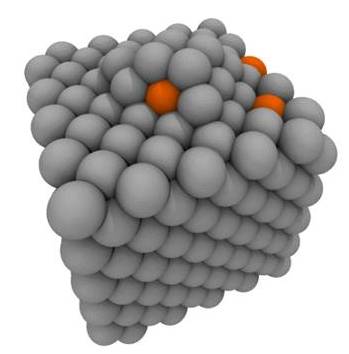
Accelerated molecular dynamics method
Molecular dynamics (MD) is an accurate yet computationally expensive material simulation technique. A major challenge arises while studying rare-event atomic processes that occur at large time-scales while rare events are being studied. We employ different variations of accelerated MD based on original developements by Arthur Voter at Los Alamos National Lab. The figure below shows an example of a many-atom process observed with accelerated MD.
We are investigating the kinetics and thermodynamical aspects of semiconductor alloy materials. More information will appear soon.

We are currently investigating transport of ions through fuel cell electrolytes. More information will appear shortly.

We are currently investigating transport of ions/atoms in lithum ion battery electrodes. More information will appear shortly.
We are currently investigating formation of self-assembled nanostructures structures.

Here is an example of a phase diagram obtained when long-ranged repulsive and short-range attractive interactions are present.
