Primary Area of Research: Computational Chemistry
Our current research focuses on modeling chemical reactions in condensed matter systems using molecular dynamics techniques.
Our group developed novel
ab initio and QM/MM MD techniques and enhanced sampling methods to improve the accuracy and efficiency of computational methods for mechanistic studies and free energy calculations of complex catalytic systems.
We also co-develop the CPMD density functional program package. Our lab is also developing a new electronic structure program package, which will be released by the end of 2022.
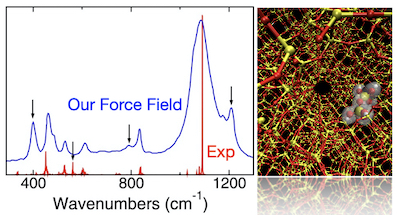
Our group has also developed atomistic force fields to model systems like polymer-oxide interface, alumino-silicates, high-performance polymers, and polymer nanocomposites.
Some of the major research achievements are below:
- Molecular Dynamics with DFT at a Higher Rung of Accuracy
Molecular dynamics simulations using the density functional theory (DFT) method have been widely used to study chemical reactions and structural and dynamic properties of molecular systems. These calculations have been done at the Generalized Gradient Approximation (GGA) level for several decades because simulations at the next higher level of theory, i.e., using hybrid density functionals, are unrealistic due to the enormous computational overhead involved. We developed a new method to speed up hybrid density functional-based molecular dynamics calculations by one order of magnitude without compromising accuracy. With this, the molecular dynamics simulation community can now model complex chemical reactions such as enzymatic reactions with a more accurate level of theory.
Relevant Publications:

-
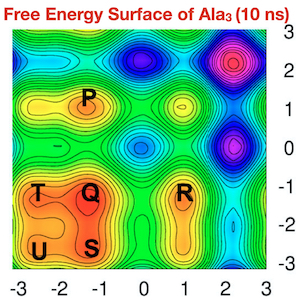
Development of New Enhanced Sampling Methods for Efficient Free Energy Calculations of Chemical Reactions
Enhance sampling techniques are widely used in molecular dynamics simulation to accelerate the barrier crossing events (such as conformational changes, chemical reactions). Such methods are also used in computing free energies along a certain set of coarse-grained coordinates or collective variables (CVs). Widely used techniques like umbrella sampling and metadynamics are limited to enhanced sampling and free energy calculations with few CVs. To overcome this limitation, we have developed methods that outperform the existing techniques and provide a way to quickly achieve converged free energy estimates and rigorous sampling of CVs. These techniques are named Temperature Accelerated Sliced Sampling (TASS) and Well-Sliced Metadynamics. The directional/controlled sampling feature of these techniques is handy for modeling complex enzymatic reactions, protein folding, drug binding, etc. The efficiency of these techniques has been further improved by using a mean-force variant of the method and by combining with replica exchange molecular dynamics techniques.
Relevant Publications:
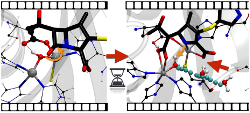
- Probing the Molecular Mechanism of Antibiotic Resistance by β-Lactamases
β-Lactamase, a bacterial enzyme, can hydrolyze nearly all the widely used β-lactam drugs efficiently, posing a significant concern to public healthcare. The antibiotic resistance originated from different classes (class-A, B, C, and D) of β-lactamases is yet to be understood in great detail. Our interest is to investigate the hydrolysis mechanism β-lactam drugs by various classes of β-lactamases using molecular dynamics techniques. Our detailed studies on various drugs and inhibitors have given hints to develop new inhibitors against β-lactamases.
Relevant Publications:
- GTP Hydrolysis Reactions by GTPases
GTP hydrolysis reactions by GTPases are of great significance due to their central role in biological regulations. For the extensively studied Ras and EF-Tu GTPases conserved glutamine or histidine is identified to be crucial for GTP hydrolysis. A group of GTPases termed HAS (Hydrophobic Amino-acid Substituted)-GTPases naturally possess a hydrophobic residue in place of Gln/His and yet efficiently hydrolyze GTP. Despite structural and biochemical studies on HAS-GTPases, their catalytic mechanism is not well understood. For most of these HAS-GTPases, catalytically crucial residues are not identified by experimental studies. We employ molecular dynamics simulations combined with metadynamics and other enhanced sampling techniques for understanding the GTP hydrolysis mechanism in HAS-GTPases. Our study is focused on two HAS-GTPases, namely Era and FeoB. These are bacterial HAS GTPases, which can be potential targets for the development of antimicrobial drugs. Therefore, understanding their catalytic mechanism is highly significant from a biological as well as therapeutic perspective. We also aim at extrapolating their mechanisms to other HAS-GTPases using structural bioinformatics.
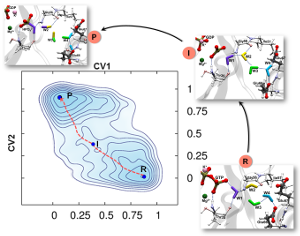
Relevant Publications:
- Development of CPMD-GULP QM/MM Interface and Reactions in Zeolites
We develop QM/MM techniques to simulate large heterogeneous catalytic systems, such as zeolite-supported metal clusters. We interfaced the plane wave DFT-based CPMD code with molecular mechanics code GULP. We investigated reactions of alkenes catalyzed by Rh clusters supported in a cavity of
Y-zeolite using our developed CPMD/GULP QM/MM interface program.
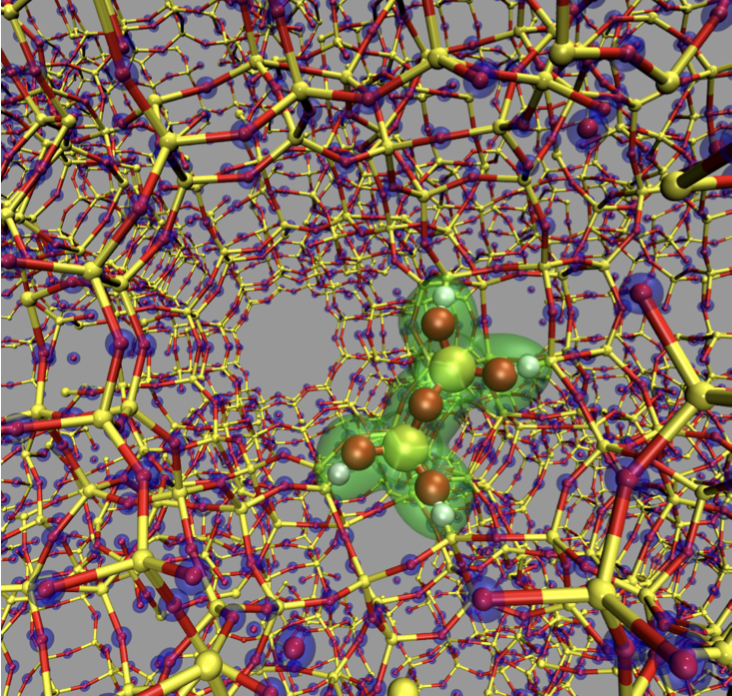 Currently, we are exploring other zeolite types and their catalytic activity.
Currently, we are exploring other zeolite types and their catalytic activity.
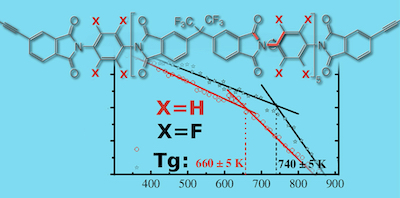
- Oxidative Degradation of High-Temperature Polymers
High-performance polymers (HPPs) have immense applications in the aerospace and electronics industries owing to their high thermal stability and resistance to various environmental conditions. The growing requisite for materials having high thermo-oxidative stability makes the design and development of these materials an active area of research. Improving the high-temperature long-term thermo-oxidative stability of high-performance polymers is crucial for their applications, particularly aerospace applications. For designing novel polymers with improved thermo-oxidative stability, it is vital to identify the most reactive moiety of the polymer toward oxidation and understand the oxidation mechanism. In this project, the thermo-oxidative degradation of many HPPs is investigated using quantum chemical and microkinetic studies. A database containing the rate constants of various oxidation pathways and a visual interface is also developed.