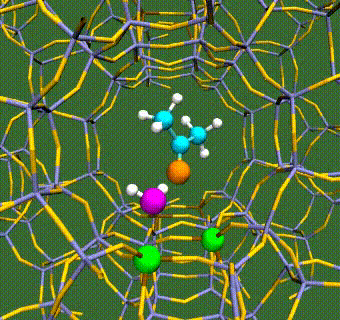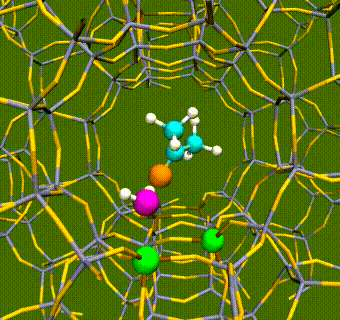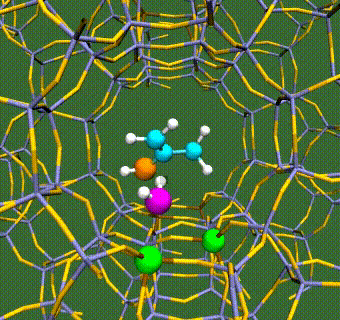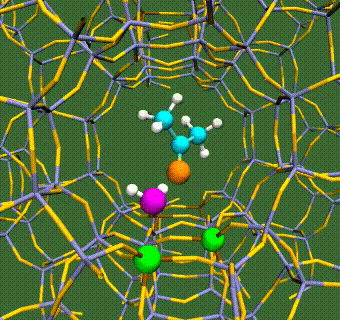
Abstract: Herein, we have performed DFT+U calculations to investigate the adsorption and activation of CO2 on a Ta-doped anatase TiO2(101) and compared it to that on a pristine anatase TiO2(101). Replacing Ti with a higher valence Ta-dopant results in an excess electron, which localizes on the neighboring Ti atom. We find that there is a slight increase in adsorption energy of CO2 with Ta-doping, however, there is no significant change in the barriers for CO2 activation. Further, presence of a water molecule enhances the adsorption energy and stabilizes the CO2 molecule having both linear and bent configurations (activated). This stabilization is more pronounced for a activated CO2 molecule, which is attributed to the strong H-bonding. Apart from being a reactant, water also acts as a co-catalyst and significantly reduces the barrier for CO2 activation. This is true for both anatase TiO2(101) and Ta-doped anatase TiO2(101). We have also investigated the activation of CO2 via hydrogen and two electron transfer, which constitutes the limiting step in the photochemical reduction of CO2 to CH4. Our calculations show that the barriers for CO2 activation via the two-electron pathway are similar for both anatase TiO2(101) and Ta-doped TiO2(101). However, presence of a water molecule on Ta-doped TiO2(101), not only stabilizes the CO2 molecule but also reduces the barrier for CO2 activation dramatically by acting as a co-catalyst.



Abstract: Computations based on density functional theory are carried out to examine the mechanism of photocatalytic oxidation of methane to methanol with H2O2 as oxidant and water as co-catalyst over anatase TiO2 (101) surface. The reaction proceeds with hydrogen abstraction from methane followed by the formation of surface methoxy species, which is reduced to methanol. We compare the reaction energetics for C-H dissociation in the presence and absence of surface defect, but find no discernible impact of O-vacancy on methane oxidation. In comparison, hydroxyl produced as a result of H2O2 or H2O photo-decomposition dramatically reduces the barrier for CH3-H bond cleavage. The reaction proceeds further by the reduction of surface methoxy group and is the rate-limiting step for methanol formation. Additionally, we find that methyl can also react with water to form methanol with a considerably lower barrier, suggesting an active involvement of water for methanol formation. We also study the role of water as a co-catalyst and observe significant reduction in barriers, facilitated by alternate pathway for proton transfer. The reaction pathways presented provide valuable in- sights into the mechanism of methane oxidation in the presence of H2O2 as oxidant and demonstrate the rate-enhancing role of water for these steps.
Abstract: Recently remarkably high ionic conductivities (up to 5.5 x 10-3 Ω-1cm−1) at 25 °C have been achieved in sodium superionic conducting (NASICON) solid-state electrolytes via aliovalent cation doping. Doping in NASICON leverages the synergic effects of bottleneck opening (lattice expansion) and correlated migration (higher carrier concentration). However, there is no report published that isolates the effect of correlated migration induced by heterovalent doping without the influence of structural/bottleneck changes. Therefore, here we report for the first time the substitution of trivalent Ru3+ cation into the Zr4+ site to enhance the ionic conductivity of the NASICON electrolyte. Even though Ru3+ has a smaller ionic radius than Zr4+ and leads to bottleneck shrinkage, high ionic conductivity is achieved on doping Ru as compared to undoped NZSP. Using first-principles calculations, we show that even though the bottleneck for Na+ migration shrinks, the diffusion barriers are nearly the same owing to the weaker Na-framework interaction. The doped electrolyte showed an ionic conductivity of 2.1 x 10-3 Ω-1cm−1, an ionic transference number of ∼ 98.9 %, and exhibited stable sodium plating/stripping at 0.5 mA-cm−2 for 100 h with an overpotential of 20 mV owing to the fused microstructure. The assembled all-solid-state battery with Ru-doped electrolyte exhibited an outstanding initial capacity of 87 mAh-g−1 at 0.3C as compared to 70 mAh-g−1 with undoped electrolyte. Compared to the pristine sample, which had capacity retention of 60 % after 100 cycles, the cell containing Ru-doped electrolyte demonstrated stable cycling with an 87 % capacity retention without external pressure.
Abstract: We use quantum chemical calculations to examine methane pyrolysis catalyzed by molten sodium. Such a reaction is performed in a bubble column filled with molten catalyst. The ascending bubble contains methane and sodium vapor where the reaction takes place. Therefore, the reaction is catalyzed by the liquid surface of the bubble, by the sodium vapor in the bubble and in the headspace above the melt. We examined here the rate of 24 possible reactions in the gas and concluded that the gas phase pyrolysis is ineffective. On the other hand the liquid surface is as good a pyrolysis catalyst as any of the melts studied so far.
Abstract: Supported molybdenum oxides have been widely employed as catalysts for variety of reactions including upgrading of bio-oil via hydrodeoxygenation. Herein, we perform spin-polarized density functional theory calculations to investigate the structures and reactivity of monomeric MoO species supported on ZrO in the absence and presence of hydroxyl(s). Monomeric molybdenum moieties were found to have a mono-oxo (one molybdenyl), di-oxo (two molybdenyls) or a pero-oxo (one molybdenyl and Mo coordinated to a peroxide) configuration. Based on the formation energies, we find that mono-oxo moieties are the most stable among the three. This is also true for the moieties in the presence of hydroxyls on the support. Further, the computed vibrational frequencies of a mono-oxo moiety on a hydroxylated support is a closer match to the peaks observed in experiments. A thermodynamic analysis of the molybdenum oxide moieties on a hydroxylated support indicated that the dehydoxylation is favoured at high temperatures and low partial pressures of water. On adsorption of hydrogen on the oxygen of the support, hydrogen loses its electron and reduces Mo of the monomeric moiety without making an oxygen vacancy. We studied the activity of this reduced site for deoxygenation of anisole to benzene. The overall barriers for this reaction are comparable to previously computed barriers for deoxygenation of bio-oil model compounds on an oxygen-vacant molybdenum oxide surfaces, suggesting that this may be an important mechanistic pathway during a hydro-deoxygenation process.
Abstract: A melt containing 1% FeCl3 and 99% KCl is a good catalyst for methane pyrolysis, while molten KCl has almost no catalytic activity. We use ab initio molecular dynamics, based on density functional theory, to examine why this surprising behavior takes place. We find that in this ionic salt, the charge on the iron fluctuates between Fe3+ and Fe2+, and the conversion to Fe2+ takes charge from several chlorine ions, which become active for methane dissociation. We speculate that such activation is general and takes place for any dopant Mn+, which has a state M(n–1)+.
Abstract: Aldol condensation of furfural with acetone is an important biofuel upgrading reaction. Herein, we investigated the rate-controlling step of aldol condensation, i.e., the keto–enol tautomerization of acetone, in Sn-doped BEA (Sn-BEA) and hydrogen-compensated Al-doped BEA (H-BEA) zeolites. We also investigated the rate-enhancing role of molecular water at 373 K. In dehydrated H-BEA, we find that the reaction takes place concertedly with a free-energy barrier, which is ∼160 kJ/mol lower than the uncatalyzed gas-phase reaction. The barrier is further reduced in the presence of water, which provides an alternate path for proton transfer. Further, we find that defect-free Sn-BEA under dehydrated conditions is catalytically inactive for the tautomerization of acetone. In the presence of H2O, we modeled two different active sites: monohydrated closed Sn-site and an open Sn-site formed by partial hydrolysis of a closed Sn-site. We find that a monohydrated closed Sn-site is thermodynamically more stable than an open Sn-site. Our calculations suggest that both configurations have comparable activity toward keto–enol tautomerization of acetone and are much higher than a dehydrated closed Sn-site. Overall, our calculations suggest that the activity of BEA zeolite is enhanced in the presence of low concentrations of water, which acts as a cocatalyst. This is true for both Sn-doped and Al-doped BEA, and the effect is more dramatic in the Sn-doped BEA.
Abstract: The application of Raman spectroscopy to heterogeneous catalysts has undergone several changes over the years. Based on the information collected during characterization of the catalysts, these changes can be categorized into the three generations of applications. In the first-generation of applications, information from Raman spectroscopy is used to characterize the catalysts when the environment surrounding the catalyst is not well controlled, for example, ambient conditions. This is also referred to as ex situ characterization. The second-generation applications are those where the vibrational information from the catalysts under controlled environments or in situ studies is examined, and the nature of the solid catalyst and gas-solid catalyst interactions are deciphered. Finally, the third-generation applications involve the simultaneous use of in situ Raman spectroscopy and reaction data to directly correlate the structure and reactivity information of the working heterogeneous catalysts. Such studies are referred to as operando Raman spectroscopy.More recently, Raman spectroscopy has been coupled with other characterization techniques to obtain additional information simultaneously, which are then correlated with the reactivity data. Furthermore, computational techniques coupled with operando Raman and other techniques are becoming in vogue for a more comprehensive understanding of the catalytic system. In this chapter, we primarily take up two case studies. In one, we highlight the three generations of application of Raman spectroscopy, and in the other, we show the importance of using density functional theory coupled with Raman spectroscopy to identify the molecular nature of the catalyst. The proper applications of Raman spectroscopy to heterogeneous catalysis is faced with several challenges. However, the future looks bright, and it is expected that we will continue to see a significant increase in research in this exciting area.
Abstract: We use a simple generic model to study the desorption of atoms from a solid surface in contact with a liquid, by using a combination of Monte Carlo and molecular dynamics simulations. The behavior of the system depends on two parameters: the strength ϵLS of the solid-liquid interaction energy and the strength ϵLL of the liquid-liquid interaction energy. The contact with the solid surface modifies the structure of the adjacent liquid. Depending on the magnitude of the parameters ϵLS and ϵLL the density of the liquid oscillates with the distance from the surface for as far as five atomic layers. For other values of the parameters the density of the liquid near the surface is much lower than that of the bulk liquid, a process sometimes called dewetting. To describe the desorption rate we have determined the number N(t) of atoms that were adsorbed initially and are still adsorbed at time t. The average of this quantity decays exponentially and this allows us to define a desorption rate coefficient kd. We found that kd satisfies the Arrhenius relation. The logarithm of the pre-exponential is a linear function of the activation energy (the so-called compensation effect). We have also examined two approximate mean-field like theories of the rate constant: one calculates the activation free energy for desorption; the other uses transition state theory applied to the potential of mean force. Both methods fail to reproduce the exact desorption rate constant kd. We show that this failure is due to the presence of substantial recrossing of the barrier (which introduces errors in transition state theory) and the presence of large fluctuations in the desorption rate of individual molecules whose effect is not captured by mean-field theories.
Abstract: Reduced molybdenum oxides are versatile catalysts for deoxygenation and hydrodeoxygenation reactions. In this work, we have performed spin-polarized DFT calculations to investigate oxygen healing energies on reduced molybdenum oxides (reduced α-MoO3, γ-Mo4O11 and MoO2). We find that Mo+4 on MoO2 (100) is the most active for abstracting an oxygen from the oxygenated compounds. We further explored CO2 adsorption and dissociation on reduced α-MoO3 (010) and MoO2 (100). In comparison to reduced α-MoO3 (010), CO2 adsorbs more strongly on MoO2 (100). We find that CO2 dissociates on MoO2 (100) via a two-step process, the overall barrier for which is 0.6 eV. This barrier is 1.7 eV lower than that on reduced α-MoO3 (010), suggesting a much higher activity for deoxygenation of CO2 to CO. As H2 dissociation is shown to be the rate-limiting step for hydrodeoxygenation reactions, we also studied activation barriers for H2 chemisorption on MoO2 (100). We find that the chemisorption barriers are 0.7 eV lower than that reported on reduced α-MoO3 (010). Finally, we have studied the dissociation (C−O cleavage) of anisole (a lignin-based biofuel model compound) on MoO2 (100). We find that anisole binds very strongly on MoO2 (100) with an adsorption energy of −1.47 eV. According to Sabatier's principle, strongly adsorbing reactants poison the catalyst surface, which may explain the low activity of MoO2 observed during experiments for hydrodeoxygenation of anisole.
Abstract: We have performed a combined quantum mechanical and microkinetic modeling study to understand the nascent decomposition pathways of methane pyrolysis, catalyzed by gas-phase ZnCl2, in a constant pressure batch reactor at 1273 K. We find that ZnCl2 catalyzes methane pyrolysis with an apparent activation energy of 227 kJ/mol. We have also performed sensitivity analysis on a reaction network comprising initiation, termination, and primary propagation reactions. The results suggest that the whole reaction network can be simplified to four reactions, which contributes to the initial rate of methane decomposition. Based on these insights, we have also explored the catalyzing effects of gas-phase AlCl3, CoCl2, CuCl2, FeCl2, and NiCl2 for methane decomposition. Our calculations suggest that gas-phase CuCl2 and NiCl2 are the most active catalysts among the metal halides studied in this work.
Abstract: We employ ab initio density functional theory calculations to investigate the mechanisms of phenol (a model compound for lignin-derived bio-oil) alkylation with propylene to 2-isopropylphenol, in the presence and absence of water on H-BEA. We also compute the reaction pathways for phenol alkylation in the uncatalyzed gas phase to understand H-BEA catalysis. In the uncatalyzed gas phase, phenol acts as a self-catalyst, activating propylene by transferring a proton from phenolic oxygen and facilitating the carbon–carbon bond formation reaction. We find that the lowest barrier process proceeds via an intermediate both in the uncatalyzed gas phase and on dry H-BEA. On H-BEA, the intermediate is stabilized by the transfer of a zeolitic proton. Our calculations show that, in comparison to the uncatalyzed gas phase, H-BEA reduces the barrier for converting the reactant complex to the intermediate by ∼40 kJ mol−1, which translates to a four order-of-magnitude increase in the transition-state theory rate coefficient at 350 °C. However, we find that the rate coefficient for the next step, which is also the rate-limiting step, remains unchanged on H-BEA. We further find that the presence of water reduces the activation barrier for this step—i.e., the rate-limiting step—by ∼30 kJ mol−1, which leads to a two order-of-magnitude increase in the transition-state theory rate coefficient at 350 °C.
Abstract: In this study, we have modelled the non-enzymatic hydrolysis of ATP in the gas-phase and the aqueous-phase by performing ab initio molecular dynamics simulations combined with an enhanced sampling technique. In the gas-phase, we studied hydrolysis of fully protonated ATP molecule, and in the aqueous-phase, we studied hydrolysis of ATP coordinated with: a) two H+ ions (H-ATP), b) Mg2+ (Mg-ATP), and c) Ca2+ (Ca-ATP). We show that gas-phase ATP hydrolysis follows a two-step dissociative mechanism via a highly stable metaphosphate intermediate. The Adenine group of the ATP molecule plays a crucial role of a general base; temporarily accepting protons and thus helping in the elimination-addition process. In the aqueous-phase hydrolysis of ATP, we find that the cage of solvent molecules increases the stability of the terminal phospho-anhydride bond through a well-known cage-effect. The nature of the ions has an important effect on the mechanism of the reaction. We find a two-step dissociative-type mechanism for H-ATP, a single-step dissociative-type mechanism for Mg-ATP, and an SN-2 type concerted hydrolysis pathway for Ca-ATP.
Abstract: We have performed exact classical rate calculations to compute adsorption and desorption rate constants with a model representative of a real system. We compute the desorption rate using transition-state theory by taking the dividing-surface far from the surface of the solid. We find that using a mean-field assumption, i.e., applying potential of mean force to transition state theory, could lead to two orders-of-magnitude errors in the rate constant owing to large fluctuations in the desorption barrier. Furthermore, we compute the adsorption rate by including a dynamical factor which reflects the probability of sticking to the solid surface. We find that the sticking probability is highly sensitive to the coverage. Also, we find that the adsorption rate computed from the mean-field assumption is not very different from the exact adsorption rate. We also compute entropic contribution to desorption rates and compare it to that obtained from two limiting models of adsorption—2D ideal gas and 2D ideal lattice gas. We show that at high temperatures (700 K), the entropic contribution to desorption rates computed from the exact calculations is very close to that obtained from the 2D ideal gas model. However, for lower to intermediate temperatures from 200 K to 500 K, the entropic contributions cover a wide range which lies in between the two limiting models and could lead to over two-orders-of-magnitude errors in the rate coefficient.
Abstract: We review here some aspects of computational work on the catalytic chemistry of oxides. The difficulties of using density functional theory in calculations are explained. Different ways of structural or chemical modifications aimed at improving catalytic activity are reviewed. The reaction mechanism of partial oxidation reaction catalyzed by oxides is discussed. The focus is on qualitative design rules rather than on obtaining highly accurate computational results.
Abstract: This chapter examines the successes and the challenges of computational design of catalysts. It explores and learns from a crude example of experimental screening for catalysts for an exothermic reaction. There are several rules that make such rapid computational screening possible: the Sabatier principle, linear-scaling, and the Brønsted-Evans-Polanyi (BEP) relation. Scaling relations can be developed for larger molecules, which make two bonds with the solid surface, through two different atoms. Oxide catalysts have numerous applications. The chapter discusses few rules discovered through computations. It illustrates many of the problems faced by most large-scale catalytic processes. A useful catalyst must be cheap to make and it should not contain expensive or rare ingredients. Another important property of a good catalyst is its resistance to poisoning. Density functional theory (DFT) is the only practical option. DFT is approximate, especially when calculating activation energies.
Abstract: Metals that are active catalysts for methane (Ni, Pt, Pd), when dissolved in inactive low–melting temperature metals (In, Ga, Sn, Pb), produce stable molten metal alloy catalysts for pyrolysis of methane into hydrogen and carbon. All solid catalysts previously used for this reaction have been deactivated by carbon deposition. In the molten alloy system, the insoluble carbon floats to the surface where it can be skimmed off. A 27% Ni–73% Bi alloy achieved 95% methane conversion at 1065°C in a 1.1-meter bubble column and produced pure hydrogen without CO2 or other by-products. Calculations show that the active metals in the molten alloys are atomically dispersed and negatively charged. There is a correlation between the amount of charge on the atoms and their catalytic activity.
Abstract:MoO3 is a versatile catalyst for oxidation reactions that consists of bilayers connected by van der Waals interaction. In principle, a MoO3 nanocrystal can be exfoliated to create two-dimensional ribbons. For this article, we study the difference between the chemistry of slabs having a variety of crystal faces and that of the edges of ribbons cut from a two-dimensional bilayer. As a descriptor of chemical reactivity we use the energy of oxygen-vacancy formation: the easier it is to form an oxygen vacancy, the better oxidant the face of a slab or the edge of a two-dimensional ribbon is. We find that the properties of ribbon edges are different from those of the corresponding slab surfaces. The surface energies of slabs are in the order (010)s < (100)s < (101)s < (001)s, whereas the edge energies of ribbons are in the order ⟨100⟩r ≈ ⟨101⟩r < ⟨001⟩r (the subscript s indicates a slab, and r, a ribbon). Among the surfaces studied, we have found that (001)s and (101)s faces have the lowest oxygen-vacancy formation energies, and (010)s has the highest. In contrast, among the edges studied, ⟨101⟩r has the lowest vacancy formation energies. Our calculations suggest that no benefit is obtained by creating ⟨100⟩r or ⟨001⟩r ribbon edges. However, a significant decrease of oxygen-vacancy formation energies is observed on formation of ⟨101⟩r edge by exfoliating (101)s slabs. Also, among the structures studied, we found ⟨101⟩r edges to be the most reactive and (010)s surfaces to be the least reactive.
Abstract:Oxygen vacancy formation energies are often used as a descriptor of the catalytic activity of metal oxides for oxidation reactions having the Mars–van Krevelen mechanism. When these energies are calculated, it is often assumed that they depend only on the concentration of the vacancies in the top oxygen layer. Previous work has shown that in the case of TiO2 and V2O5, the energy of vacancy formation depends not only on their concentration but also on the manner in which they are distributed on the surface. However, the energy change due to the change of configuration in these systems is very small. Here, we find that in the case of α-MoO3(010) the dependence on the energy of vacancy formation of the distribution of vacancies is very large: if the lattice made by the vacancies consists of parallelograms, the energy of vacancy formation is 0.4 eV smaller than when the lattice consists of rectangles (the two systems having the same vacancy concentration).
Abstract: Density functional theory is used to determine differences in hydrogen abstraction and ammonia binding energies between two zeolites (BEA and MFI-type) and two α-quartz surfaces doped with Al, B, Sc, or Ga. One of the questions we wanted to answer is whether the fact that zeolite cages are made of a silica monolayer plays any role in their catalytic activity. We find no important difference. Doped α-quartz has acid hydroxyls such as those in zeolites; however, their density is very low, and doped quartz is not a shape selective catalyst. Therefore, the doped silica examined here is an inferior acid catalyst when compared to BEA or MFI.
Abstract:We use the Potts-lattice gas model to study nucleation at and near the eutectic composition. We use rare-event methods to compute the free energy landscape for the competing nucleation products, and short trajectories at the barrier top to obtain prefactors. We introduce a procedure to tune the frequency of semigrand Monte Carlo moves so that the dynamics of a small closed system roughly resemble those of an infinite system. The non- dimensionalized nucleation rates follow trends as predicted by the classical nucleation theory. Finally, we develop corrections that convert free energy surfaces from closed (canonical) simulations into free energy surfaces from open (semigrand) simulations. The new corrections extend earlier corrections to now address situations like nucleation at the eutectic point where two products nucleate competitively.
Abstract: This chapter focuses on a frontier for molecular simulation: nucleation of solute precipitates from solution. The chapter refers to simulations of solute precipitate nucleation with a fixed number of solute and solvent molecules simulations of “closed systems”. Two simple cases of heterogeneous nucleation are considered: (i) a spherical cap model for nucleation on a hard flat surface and (ii) a lens model for nucleation at fluid–fluid interfaces. Over the decade following the discovery of two'step nucleation, several computational studies have pointed to two'step nucleation as a more general phenomena occurring even at points where there is no metastable fluid–fluid critical point. Rare events simulation methods can relax many of the assumptions made by classical nucleation theory (CNT). The chapter concludes with case studies on laser'induced nucleation, and on nucleation of methane hydrates and calcium carbonate.
Abstract:We modeled nascent decomposition processes in cellulose pyrolysis at 327 and 600 °C using Car–Parrinello molecular dynamics (CPMD) simulations with rare events accelerated with the metadynamics method. We used a simulation cell comprised of two unit cells of cellulose Iβ periodically repeated in three dimensions to mimic the solid cellulose. To obtain initial conditions at reasonable densities, we extracted coordinates from larger classical NPT simulations at the target temperatures. CPMD-metadynamics implemented with various sets of collective variables, such as coordination numbers of the glycosidic oxygen, yielded a variety of chemical reactions such as depolymerization, fragmentation, ring opening, and ring contraction. These reactions yielded precursors to levoglucosan (LGA)—the major product of pyrolysis—and also to minor products such as 5-hydroxy-methylfurfural (HMF) and formic acid. At 327 °C, we found that depolymerization via ring contraction of the glucopyranose ring to the glucofuranose ring occurs with the lowest free-energy barrier (20 kcal/mol). We suggest that this process is key for formation of liquid intermediate cellulose, observed experimentally above 260 °C. At 600 °C, we found that a precursor to LGA (pre-LGA) forms with a free-energy barrier of 36 kcal/mol via an intermediate/transition state stabilized by anchimeric assistance and hydrogen bonding. Conformational freedom provided by expansion of the cellulose matrix at 600 °C was found to be crucial for formation of pre-LGA. We performed several comparison calculations to gauge the accuracy of CPMD-metadynamics barriers with respect to basis set and level of theory. We found that free-energy barriers at 600 °C are in the order pre-LGA < pre-HMF < formic acid, explaining why LGA is the kinetically favored product of fast cellulose pyrolysis.
Abstract:We have modeled the transformation of cellulose Iβ to a high temperature (550 K) structure, which is considered to be the first step in cellulose pyrolysis. We have performed molecular dynamics simulations at constant pressure using the GROMOS 45a4 united atom forcefield. To test the forcefield, we computed the density, thermal expansion coefficient, total dipole moment, and dielectric constant of cellulose Iβ, finding broad agreement with experimental results. We computed infrared (IR) spectra of cellulose Iβ over the range 300–550 K as a probe of hydrogen bonding. Computed IR spectra were found to agree semi-quantitatively with experiment, especially in the O–H stretching region. We assigned O–H stretches using a novel synthesis of normal mode analysis and power spectrum methods. Simulated IR spectra at elevated temperatures suggest a structural transformation above 450 K, a result in agreement with experimental IR results. The low-temperature (300–400 K) structure of cellulose Iβ is dominated by intrachain hydrogen bonds, whereas in the high-temperature structure (450–550 K), many of these transform to longer, weaker interchain hydrogen bonds. A three-dimensional hydrogen bonding network emerges at high temperatures due to formation of new interchain hydrogen bonds, which may explain the stability of the cellulose structure at such high temperatures.
Abstract:We have studied base strengths of nitrogen-substituted (nitrided) zeolites with faujasite (FAU) structure by calculating sorption energies of probe molecules (BF3 and BH3) using density functional theory with mixed basis sets applied to embedded clusters. BH3 was found to be a better probe of base strength because it does not introduce competing metal−fluorine interactions that obfuscate trends. In all cases, the base strengths of nitrided zeolites (denoted M−N−Y) were found to exceed those of the corresponding standard M−Y zeolites, where M = Li, Na, K, Rb, or Cs charge-compensating cations. We have found that for a particular Si:Al ratio, BH3 sorption energies vary in the order Li < Na < K ∼ Rb ∼ Cs. Sorption energy and hence base strength was found to decrease with increasing Si:Al ratio from 1 to 3 beyond which the base strength was found to increase again. The initial regime (1 < Si:Al < 3) is consistent with the prevailing understanding that the base strength increases with Al content, while the latter regime (Si:Al > 3) involves the surprising prediction that the base strength can be relatively high for the more stable, high-silica zeolites. In particular, we found the sorption energy in Na−N−Y (Si:Al = 11) to be nearly equal to that in (Si:Al = 1). Taken together, these results suggest that K−N−Y (Si:Al = 11) optimizes the balance of activity, stability, and cost.
Abstract: We have modeled the formation kinetics of nitrogen-substituted (nitrided) zeolites HY and silicalite; we have also modeled the stability of nitrided sites to heat and humidity. These kinetic calculations are based on mechanisms computed from DFT-computed pathways reported in our previous work. Reactant ammonia and product water concentrations were fixed at various levels to mimic continuous nitridation reactors. We have found that zeolite nitridation — replacing Si–O–Si and Si–OH–Al linkages with Si–NH–Si and Si–NH2–Al, respectively — proceeds only at high temperatures (> for silicalite and > for HY) due to the presence of large overall barriers. These threshold temperatures are in good agreement with experiments. Nitridation yields were found to be sensitive to water concentration, especially for silicalite where nitridation is more strongly endothermic. As a result, overall nitridation yields in silicalite are predicted to be much lower than those in HY. The stability of nitrided sites was investigated by modeling the kinetics of nitridation in reverse, going back to untreated zeolite plus ammonia. Using 10 h as a benchmark catalyst lifetime, nitrided silicalite and HY half-lives exceeded 10 h for temperatures below 275 and 500 °C, respectively, even at saturation water loadings. As such, our calculations suggest that nitrided silicalite and HY zeolites require high temperatures to form, but once formed, they remain relatively stable, auguring well for their use as shape-selective base catalysts.
Abstract: We have performed embedded-cluster calculations using density functional theory to investigate mechanisms of nitrogen substitution (nitridation) in HY and silicalite zeolites. We consider nitridation as replacing Si–O–Si and Si–OH–Al linkages with Si–NH–Si and Si–NH2–Al, respectively. We predict that nitridation is much less endothermic in HY (29 kJ/mol) than in silicalite (132 kJ/mol), indicating the possibility of higher nitridation yields in HY. To reveal mechanistic details, we have combined for the first time the nudged elastic band method of finding elusive transition states, with the ONIOM method of treating embedded quantum clusters. We predict that nitridation of silicalite proceeds via a planar intermediate involving a 
Abstract: Nanoporous acid catalysts such as zeolites form the backbone of catalytic technologies for refining petroleum. With the promise of a biomass economy, new catalyst systems will have to be discovered, making shape-selective base catalysts especially important because of the high oxygen content in biomass-derived feedstocks. Strongly basic zeolites are attractive candidates, but such materials are notoriously difficult to make due to the strong inherent acidity of aluminosilicates. Several research groups have endeavored to produce strongly basic zeolites by treating zeolites with amines, but to date there is no compelling evidence that nitrogen is incorporated into zeolite frameworks. In this communication, we detail synthesis, NMR spectroscopy, and quantum mechanical calculations showing that nitrogen adds onto both surface and interior sites while preserving the framework structure of zeolites. This finding is crucial for the rational design of new biomass-refinement catalysts, allowing 50 years of zeolite science to be brought to bear on the catalytic synthesis of biofuels.
Abstract: In reactive distillation (RD) one can conveniently manipulate the concentration profiles on the reactive stages by exploiting the difference in volatility of the various components. This property of RD can be advantageously used to improve the selectivity toward the desired product in case of series or series parallel reactions, and obtain a performance superior to the network of conventional reactors. In the previous work [Agarwal, V., et al., 2008. Attainable regions of reactive distillation—Part I. Single reactant non-azeotropic systems. Chemical Engineering Science, submitted for publication], we introduced representative unit models of RD to obtain the attainable regions of RD for non-azeotropic systems. In this work, we extend the approach to a system involving single binary azeotrope. Design guidelines have been formulated based on the residue curve maps, to obtain the improved attainable region with the help of these representative RD models either alone or in the form of their network.
Abstract:Reactive distillation (RD), a promising multifunctional reactor, can be used to improve the selectivity of the desired product by manipulating the concentration profiles in the reactive zone of the column. In this work, a new approach has been proposed to obtain the feasible regions of RD for the reactive systems involving single reactants, e.g. dimerization, aldol condensation, etc. Two new models namely the reactive condenser and the reactive re-boiler have been proposed. These models indicate the best location of the reactive zone in a column. Multistage versions of these models namely, reactive rectification and reactive stripping further expand the feasible region and are capable of representing the performance offered by a conventional RD unit. Several hypothetical non-azeotropic ideal systems have been extensively studied using these models and it has been shown that selectivity close to 100% is attainable over the entire range of conversion for a series as well as a combination of series and parallel reactions with positive reaction orders. Two industrially important cases of aldol condensation of acetone and dimerization of isobutylene have also been addressed using this approach. For porous catalysts, the presence of intra-particle diffusion resistance may limit the feasible region and even in the case of ideal non-azeotropic systems it may not be possible to obtain 100% selectivity. A methodology to incorporate pore diffusion effects is also illustrated.
Abstract: Aldol condensation of acetone in the presence of acid catalyst gives diacetone alcohol (DAA) as an intermediate product, which further dehydrates to give mesityl oxide (MO). By using reactive distillation (RD), one can improve selectivity toward DAA, by continuously removing it from the reactive zone and thereby suppressing the dehydration reaction. The presence of water in the reaction mixture has a predominant effect on the intrinsic reaction rates of the individual reactions. This rate-inhibiting effect of water can be advantageously used to improve the selectivity toward the desired intermediate products. The present study, through experiments and simulation, shows that introduction of water in RD can further increase the selectivity toward DAA. Batch kinetics of the reaction in the presence of water is studied, and a suitable kinetic expression is proposed. Further, the batch and continuous reactive distillation experiments are performed to assess the feasibility. The experimental results are explained with the help of an equilibrium-stage model, and the operating parameters for the desired performance are suggested based on the validated model.
Abstract: Dimerization of acetone (Ac) yields diacetone alcohol (DAA), which on further dehydration gives mesityl oxide (MO) along with various side-products. The reacting system is a combination of various series and parallel reactions. In the present work, the reaction is studied using a cation exchange resin (Amberlyst 15®) as catalyst. The effect of catalyst loading and temperature on reaction kinetics was evaluated and three models based on simplified Langmuir–Hinselwood mechanism are proposed. Aim of the work is to minimize undesired side-products and understand the effect of different parameters and operating modes on DAA:MO product ratio in reactive distillation (RD). It has been shown that the reaction when operated in a reactive rectification mode offers flexibility in the relative production rates of DAA and MO. The experimental results obtained are explained by simulation.
Abstract: Reactive distillation (RD), a combination of reaction and separation, holds the potential of giving selectivity/yield much above that offered by the conventional reactors or combination of them. It can be effectively used to improve selectivity of a reaction especially when an intermediate product is desired in series or parallel reactions. In this work, some representative model systems are introduced to obtain attainable region. A series as well as a combination of series parallel reactions (irreversible and mixture of reversible and irreversible reaction systems) were studied with the proposed models and it was found that for the single reactant ideal systems one can obtain almost 100% selectivity for a near quantitative conversion, which is much greater than that obtained in the conventional reactors like PFR and CSTR. These models were also studied for the non-ideal azeotropic systems and it was found that there is a limit to feasible region. The results obtained in the theoretical analysis are supported by experiments on aldol condensation of acetone.