Research
Biophysical Chemistry
Biophysical Chemistry
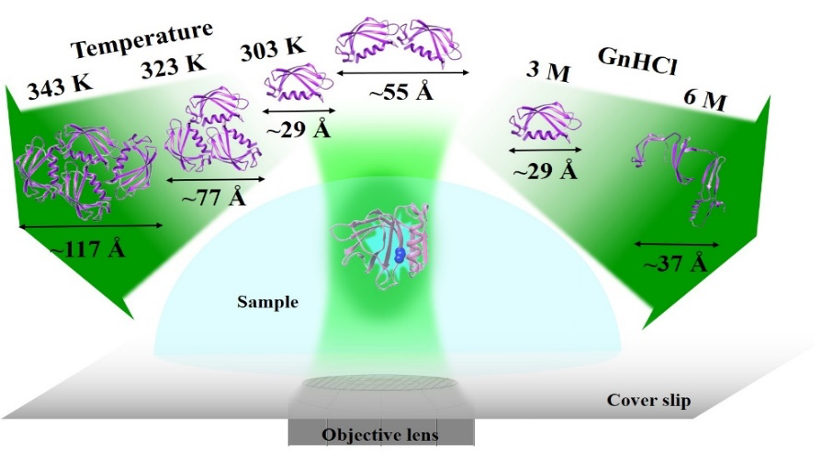
One of our main research goals is to obtain additional insight into the structural, functional and dynamical aspects of proteins. We use various ensemble average (like steady-state and time-resolved emission, CD spectroscopy) and single molecular level spectroscopic techniques (fluorescence correlation spectroscopy) routinely for this. We also use other techniques like AFM, microscopic imaging, HPLC, DLS, etc., as and when required. Another essential part of our research is the site-selective tagging of protein molecules to achieve desired specific information. Crudely, our research interest can be divided into three parts
Current research:
- We are aiming to demonstrate that, contrary to the general belief, the solvent structure (and not the positional restriction due to excluded volume) primarily determine the stability of the protein in the crowded medium
- Role of active-site dynamics in controlling the enzymatic activity
- DES (or more accurately hydrated DES) has already been postulated as the third medium inside a living system that helps to maintain physiological activities even in dehydrated conditions or at sub-freezing temperatures. This is surprising because the presence of water is the crucial factor that controls protein activity. We are aiming to have a physical insight into this
Contact:
Mechanistic insight into protein behaviour in crowded and confined media
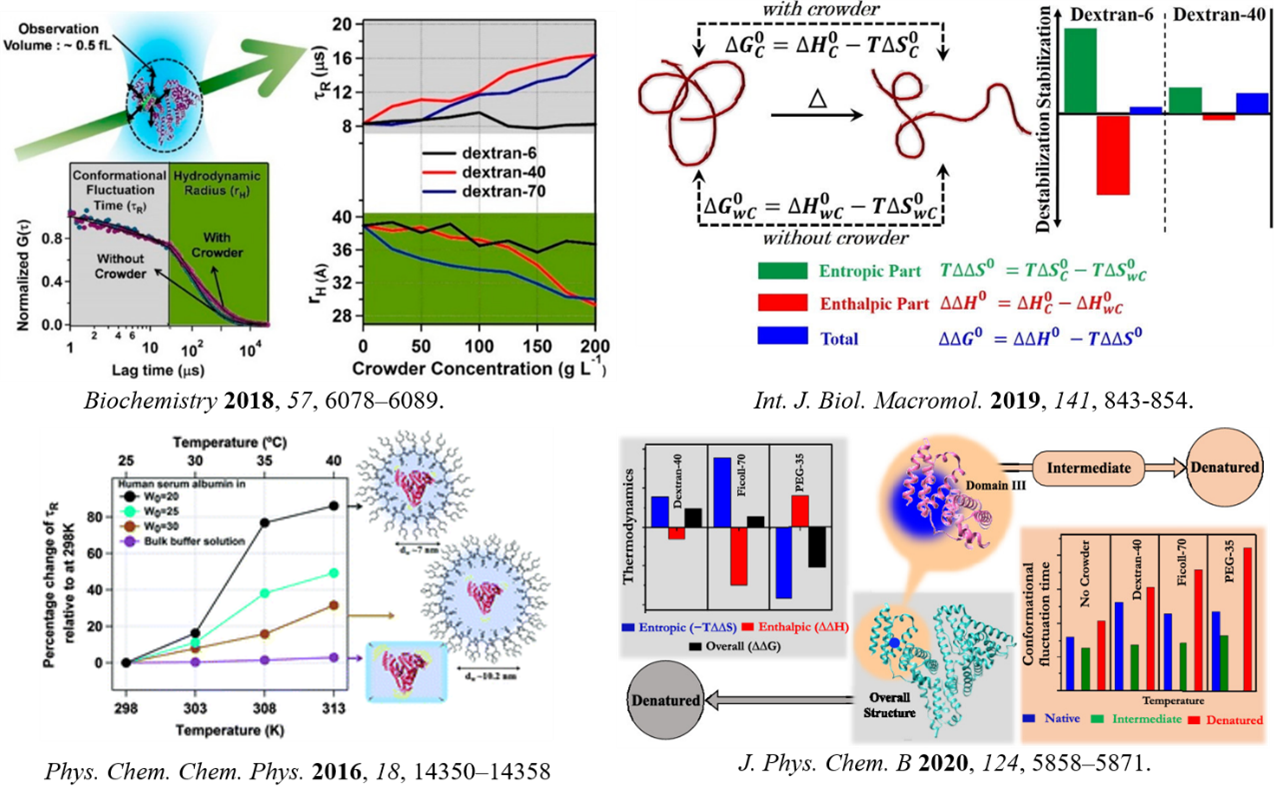
Biological functions are evolved in crowded media, and it is believed to be controlled by an interplay between the excluded volume effect (entropic) and soft interaction (enthalpic). However, many observations cannot be explained by this traditional macromolecular crowding theory. We made two fundamental claims regarding the mechanism of macromolecular crowding. We argued that the modulation in crowded milieu might also originate from the pressure exerted on the biomolecules by the crowders. We also explained the negative entropic effect by showing that the modulation of associated water structure in the presence of crowder is a critical factor for the entropic component of overall stabilization. It also explains the molecular-level basis of entropy-enthalpy compensation. (J. Phys. Chem. B 2020, 124, 5858; Int. J. Biol. Macromol. 2019, 141, 843; Biochemistry 2018, 57, 6078). In combination with the traditional crowding theory, we believe our proposition can successfully explain most of the findings that are not yet understood. We also have contemplated the active-site dynamics of papain and the effect of encapsulation within cationic and anionic reverse micelles. Our result demonstrates significant but different modulation in the two cases, highlighting how charge can be an important factor controlling protein behaviour (Spectrochim. Act. A: Mol. Biomol. Spec. 2018, 200, 202). We investigated how conformational fluctuation dynamics and solvation dynamics is modulated in the confined media. We found that conformational dynamics of domain-I of HSA become faster inside AOT reverse micelle (Phys. Chem. Chem. Phys. 2016, 18, 14350).
Active-site dynamics of protein
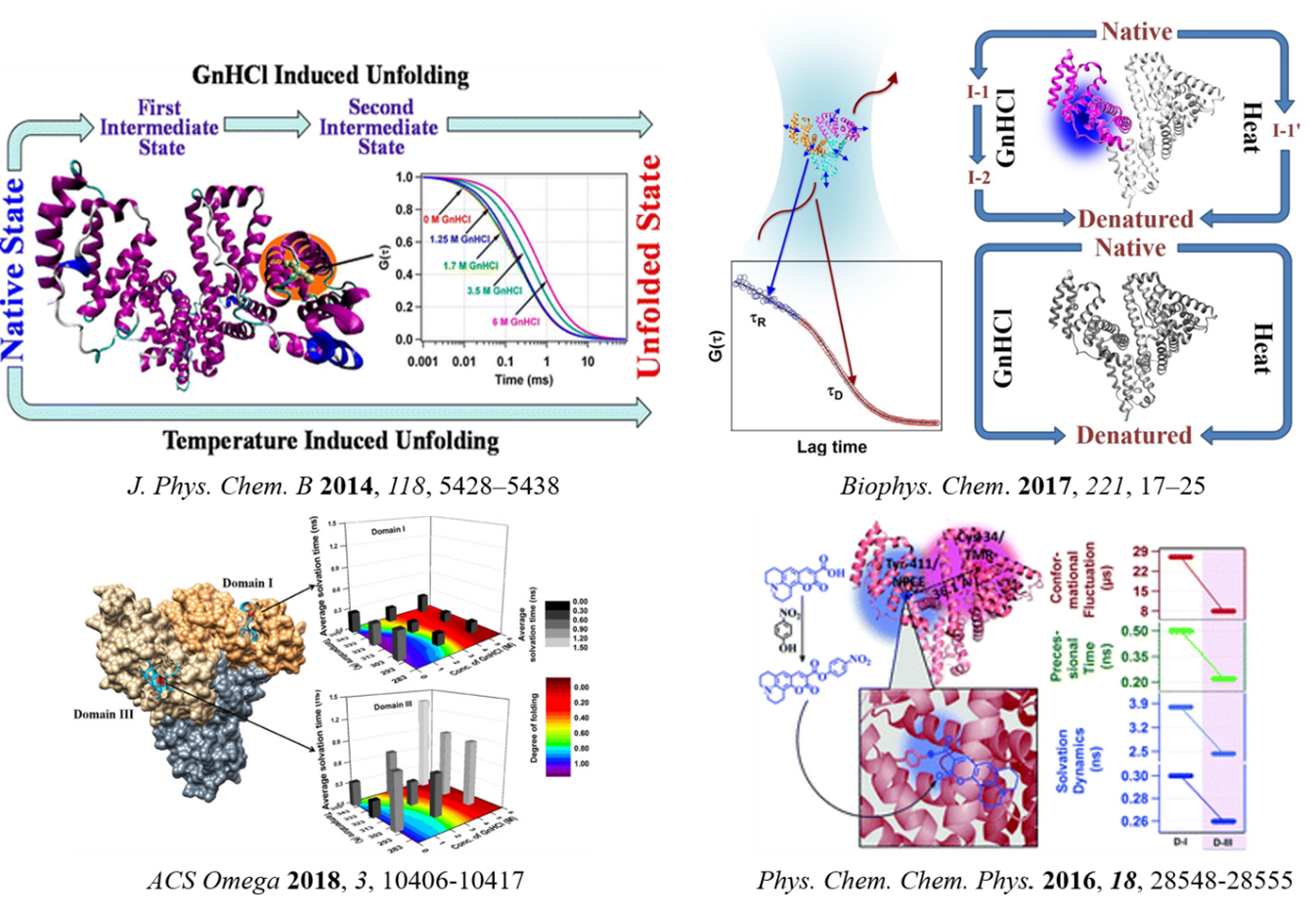
Proteins are not rigid and undergo various types of motions on different time and length scales ranging from picoseconds to minutes and 0.001 to 10 Å, respectively. Among various molecular motions of a protein molecule, side chain dynamics in the microsecond timescale are most important because its implication in enzymatic activity and signal transduction. It is also known that protein dynamics is enslaved to the associated water dynamics and the activity is largely controlled by the so-called "active site" of the protein. We contemplated active-site dynamics of various proteins along their folding-unfolding transition and aggregation pathway. By this, we observed the presence of the intermediate state(s) in the unfolding behaviour of several proteins (c.a. HSA, ?-lactoglobulin and papain), which was not detectable by ensemble average techniques (J. Phys. Chem. B 2014, 118, 5428; Biophys. Chem. 2017, 221, 17; BBA-Prot. and Proteom. 2018, 1866, 316-326). We proved that for a multi-domain protein like HSA, the solvation and rotational dynamics are different for different domains and respond differently to different perturbations (Phys. Chem. Chem. Phys. 2016, 18, 28548; ACS Omega 2018, 3, 10406). A combined solvation dynamics and fluorescence anisotropy study taking domain-I of human serum albumin (HSA) reveals that hydration dynamics plays an important role in the sucrose induced stabilization process at the low GnHCl concentration, whereas environmental restriction is responsible at the higher concentration of GnHCl (Prot. Pept. Lett. 2019, 26, 287; ACS Omega 2018, 3, 16633; Biophys. Chem. 2016, 211, 59; Protein Sci. 2013, 22, 1571).
Understanding protein behaviour in alternate media
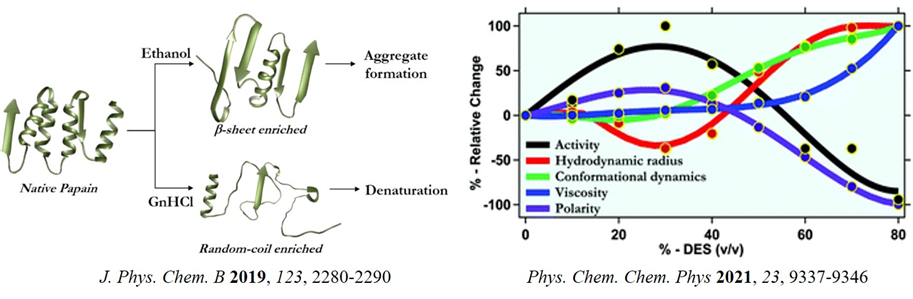
Recently, we have become interested in understanding protein behaviour in alternate media like binary solvent mixture, deep eutectic solvents and polymer matrix. Such a media has huge potential as they are environmentally benign, and has fine-tunability and biocompatibility. We scrutinized the effect of ethanol-water binary solvent mixture on an industrially important protease, papain (J. Phys. Chem. B 2019, 123, 2280). Recently, we showed that a more compact structural conformation, higher active-site flexibility, lower viscosity and higher medium polarity probably facilitate enzymatic activity in a hydrated deep eutectic solvent (DES) (Phys. Chem. Chem. Phys. 2021, 23, 9337). The gained physical insights into how enzymatic activity depends on the protein structure and dynamics and also on the physical properties of the medium might provide valuable guidelines for the rational design of DESs as biocatalytic media.
