Research
Ultrafast Spectroscopy
Ultrafast Spectroscopy
Ultrafast photochemistry and photophysics are our fundamental interest. Traditional photochemistry relies on the fact that internal conversion, vibrational cooling, and solvent relaxation are faster than the radiative transition. The study of photophysics in a timescale faster/comparable to such molecular relaxation provides unique insights. For example, photochemistry in the upper excited state manifold or involving a potential energy barrier might open up new possibilities for wavelength selective chemistry and/or reaction dynamics. We have taken up several related projects. We are basically interested in various photophysical processes e.g., photoinduced large amplitude motion, electron/proton transfer, energy transfer, excimer formation and intersystem crossing. Moreover, to mimic biological conditions, we introduce confinement. Our research can be crudely divided into the following three sub-division
Current research:
- Role of vibration in intersystem crossing process.
- Homo-energy transfer.
- Marcus inversion in excited state proton transfer reaction.
Contact:
Excited state relaxation dynamics/photophysics
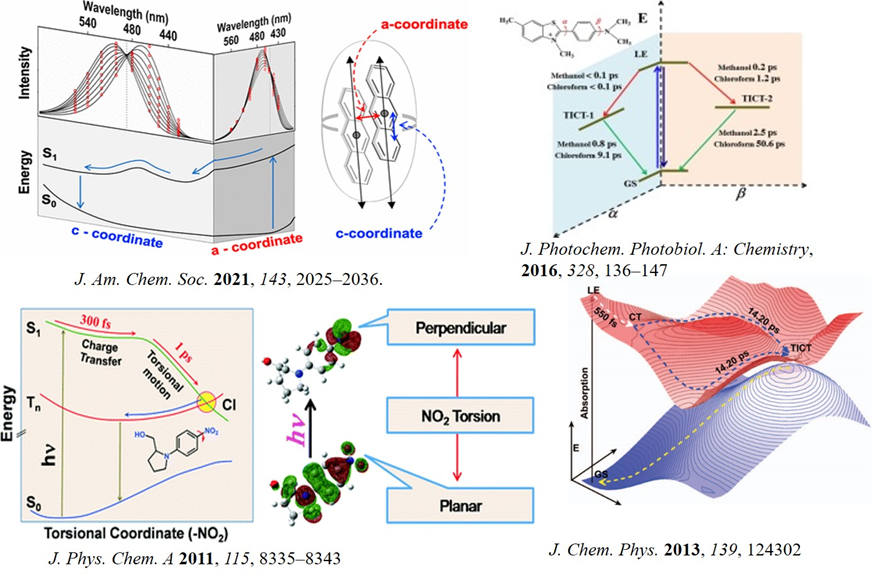
Our study on the dynamics of anthracene excimer formation under constrain shows that the excimer emission maximum is time dependent. This observation is quite unique and is not in line with the textbook examples of time-independent monomer-excimer emission of aromatics. The presence of at least one intermediate between the monomer and the excimer is inferred. It might provide insight into photodimerization reactions of organic molecules in restricted spaces offered by crystals, biological systems, and host-guest complexes (J. Am. Chem. Soc. 2021, 143, 2025-2036). In an earlier work, we postulated that the photophysics of well-known amyloid marker thioflavin-T comprises of two TICT states (one barrierless and other with a barrier) and the presence of an activation barrier in the otherwise barrierless excited state potential energy surface of auramine-O (J. Photochem. Photobiol. A: Chem 2017, 348, 287; J. Photochem. Photobiol. A: Chem, 2016, 328, 36; J. Chem. Phys. 2013, 139, 124302). Our results provide the fundamental insights into molecular relaxation dynamics that can be found useful in future. We have also explored the excited state dynamics of several GFP chromophores. Using femtosecond fluorescence up-conversion and DFT calculations we proposed a multifaceted relaxation dynamic of GFP chromophore analogues, which have importance as fluorescence marker for bioimaging. Femtosecond excited state dynamics of 4-nitrophenyl pyrrolidinemethanol provide evidence of twisted intramolecular charge transfer and intersystem crossing involving nitro group (J. Phys. Chem. A 2011, 115, 8335).
Ultrafast electron and proton transfer
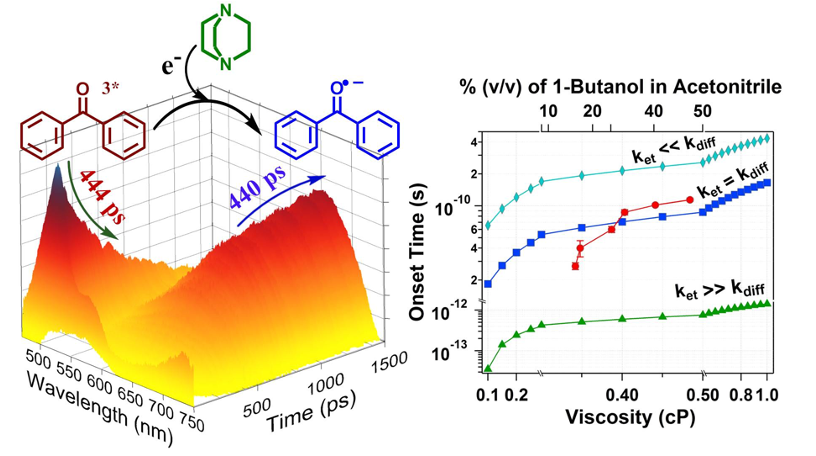
Another interest of our lab is to study fundamental electron and proton transfer reactions. Fluorescence quenching studies through steady-state and time-resolved measurements are inadequate to quantify the bimolecular electron transfer rate in bulk homogeneous solution due to diffusion constrain. We measured the true photoinduced electron transfer rate (PET) by probing the transient intermediate (J. Phys. Chem. B 2015, 119, 11253; Phys. Chem. Chem. Phys. 2017, 19, 11220). This approach is free from the constraints from diffusion. There was a long-standing debate if one could actually observe Marcus Inversion in constrained media or the observation is obscured by experimental artifacts. Our unique approach confirms that one could indeed observe Marcus Inversion in constrained media (J. Phys. Chem. B 2017, 121, 1610; J. Phys. Chem. C 2017, 121, 20205; J. Phys. Chem. A 2020, 124, 5297). A study on a model Schiff base revealed a two-step proton transfer mechanism involving the relaxation of the excited state enol form and barrier crossing from the enol to the keto form (ChemistrySelect 2018, 3, 3787).
Ultrafast solvation dynamics
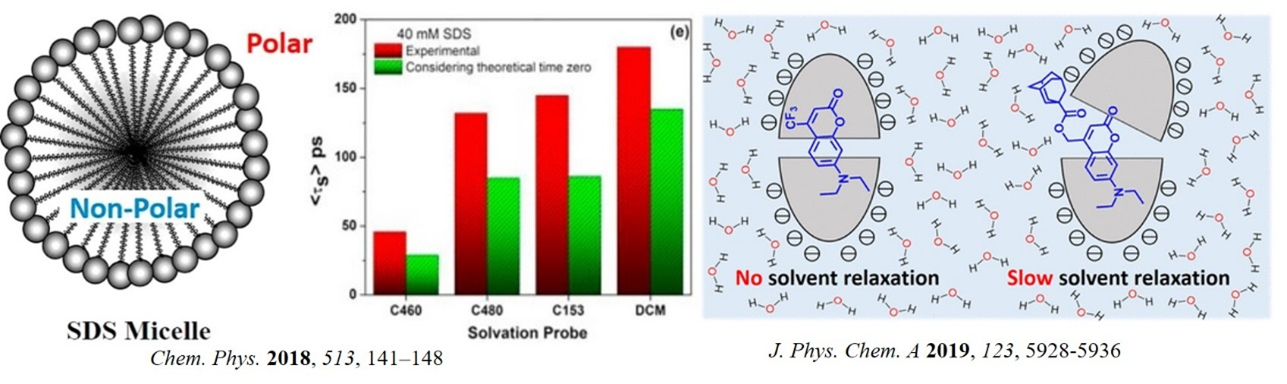
Water dynamics is a key factor that controls many fundamental reactions. Solvation dynamics in the confined environment has been a topic of interest for last three decades because it can mimic the biological environments. We investigated the probe and surfactant concentration dependence of solvation dynamics in sodium dodecyl sulfate (SDS) micelle. The average solvation times are found to follow the hydrophobicity of the probe and it was concluded that the location of the probe inside SDS micelle determines the solvation time (Chem. Phys. 2018, 513, 141). In a recent study we have also uncovered the interior environment of OA (octa acid) capsule by recording solvation dynamics of six coumarins as the fluorescent probes. Results depict that for the smaller coumarins, water cannot access the guests within the OA cavity during their excited state lifetime. However, distorted capsule structure exposes the guest to water for the larger coumarins, and a dynamics Stokes shift is observed. The average solvation time decreases with the increasing size of guests (J. Phys. Chem. A 2019, 123, 5928).
